在群体遗传学上,**基因流(也称基因迁移)**是指从一个物种的一个种群向另一个种群引入新的遗 传物质,从而改变群体“基因库”的组成。通过基因交流向群体中引入新的等位基因,是遗传变 异一个非常重要的来源,影响群体遗传多样性,产生新的性状组合。基因流会减少种群之间的差 异。
TreeMix通过从多个种群中获得等位基因频率, 返回该种群的最大似然(ML)树,并推断可能发生的杂交事件。
可以用三句话概括基本原理。 1. 使用基因频率数据可以计算出每对群体之间的协方差,这是实际的协方差 (Real value); 2. 使用基因型频率数据可以构建最大似然树,利用两个种群在树上的关系, 可以计算出协方差的估计值(Estimated value); 3. 通过实际值与估计值之间的差的大小,判断两个种群之间是否发生基因流, 即如果实际值小于估计值很多时,则说明我们构建出来的树夸大了种群之 间的差异,则说明种群之间有基因交流,因为基因流会减少种群之间的差 异。
下面所有操作基于docker run –rm -v D:\treemix:/work -it omicsclass/pop-evol-gwas:latest
### 根据LD来过滤连锁的SNP
plink --vcf filtered_snp.vcf.gz --indep-pairwise 50 10 0.2 --out tmp.ld --allow-extra-chr --set-missing-var-ids @:# --keep-allele-order
plink --vcf filtered_snp.vcf.gz --extract tmp.ld.prune.in --freq --missing --within pop.cluster.txt --out input --allow-extra-chr --set-missing-var-ids @:# --keep-allele-order
#压缩输出文件
gzip input.frq.strat
#将输出文件转换为Treemix需求的格式
/bin/python2 script/plink2treemix.py input.frq.strat.gz input_treemix.frq.gz
#假设各群体间没有基因流
treemix -i input_treemix.frq.gz -m 0 -o treemix.0 -bootstrap -k 500 -noss > treemix_0_log
Rscript script/treemix.r -i treemix -o ./ -e 0 -r pop.order.txt
#假设各群体间有一次基因流
treemix -i input_treemix.frq.gz -m 1 -o treemix.1 -bootstrap -k 500 -noss > treemix_1_log
Rscript script/treemix.r -i treemix -o ./ -e 1 -r pop.order.txt
IBD分析:
定义 :IBD(Identity By Descent),即同源相同(同源一致性,血缘一致性),指两个个体中 共有的等位基因来源于共同祖先(在两个及以上个体存在来源于同一祖先的、未发生重组的、完 全相同的DNA片段)。
由于IBD片段的能够反映个体间的遗传关系,所以IBD有非常广泛的应用。 主要有以下几个方面: 1. 估计遗传力; 2. 确定精细群体结构; 3. 检测性状变异; 4. 估计亲缘关系系数
#目录结构
## data 所需各种数据
## ibd_demo ibd示例结果(已跑完所有流程并生成全部结果)
## my_ibd 自己的ibd计算目录
## script 所需各种脚本
## software 所需beagle软件
#新建自己的ibd计算目录
mkdir my_ibd
cd my_ibd
##################################第一步 拆分染色体并分别计算IBD#################################################
#所需文件:vcf文件,chrlist:染色体名称(1列,与vcf中对应),ibd_calculate.sh
mkdir 01.chr_ibd
cd 01.chr_ibd
cp /work/ibd_demo/01.chr_ibd/ibd_calculate.sh ./
#vcf文件要过滤掉缺失位点,过滤标准至少0.8
vcftools --gzvcf xxx.vcf.gz --max-missing 1.0 --recode --stdout | gzip -c > xxx.recode.vcf.gz
#修改并运行命令calculate.sh,拆分染色体并计算ibd
nohup sh ibd_calculate.sh > ibd_calculate.sh.o &
#############################第二步 提取两个供体群体以及每个受体对应的IBD结果#################################################
#所需文件:sample_ibd.sh,ibd_pop.py,ibd_sample.py,ibd结果文件,tibd.bed文件
# listR:受体样本vcf名称(1列),listA:供体群A样本vcf名称(1列),listB:供体群B样本vcf名称(1列)
cd ../
mkdir 02.pop_ibd
cd 02.pop_ibd
#修改并运行命令 sample_ibd.sh #分别提取两个供体群的ibd值和对应的bed文件 #提取每个受体样本对应的两个供体群的bed文件,输出到新目录03.sample_ibd/下
cp /work/ibd_demo/02.pop_ibd/sample_ibd.sh ./
nohup sh sample_ibd.sh > sample_ibd.o &
###########################第三步 思路一:计算群体间rIBD 目录02.pop_ibd################################################
#所需文件(自行准备)genome.chr.ln,文件为两列,第一列是与ibd对应的染色体名称,第二列是染色体长度,与vcf中对应,见示例文件·
#命令与脚本文件:pop_ribd.sh,ribd_pop.py
#划窗口,生成bed文件
bedtools makewindows -g /work/data/genome.chr.ln -w 10000 >10K.genome.bed
#修改并运行命令 pop_ribd.sh #计算供体A群和B群分别对应的cIBD,计算两个供体群之间的rIBD
cp /work/ibd_demo/02.pop_ibd/pop_ribd.sh ./
nohup sh pop_ribd.sh > pop_ribd.sh.o &
ls
#画图
Rscript /work/script/ribd_plot.R -i ribd.txt -chr chr01 -n ribd
#必选参数:-i 输入文件 -chr 指定染色体
#可选参数:-n 输出文件前缀,-color 指定颜色, -o 输出文件目录 -x x轴标题,-y y轴标题,
# -H 生成PDF的高度(默认4),-W 生成PDF的宽度(默认6)
##################第三步 思路二:对每个受体样本计算IBD_proption 目录03.sample_ibd#################################################
#所需文件:listR:受体样本vcf名称(1列),listA:供体群A样本vcf名称(1列),listB:供体群B样本vcf名称(1列)
# 划窗口bed文件,ibd_proption.py,ibd_proption.sh
#进入上一步生成的目录,默认为03.sample_ibd/
cd ../03.sample_ibd/
cp ../02.pop_ibd/10K.genome.bed ./
cp /work/ibd_demo/03.sample_ibd/ibd_proption.sh ./
#运行命令 cibd.sh,,批量产生cIBD结果文件和最终IBD比例文件
nohup sh ibd_proption.sh > ibd_proption.sh.o &
#画图在excel中进行即可
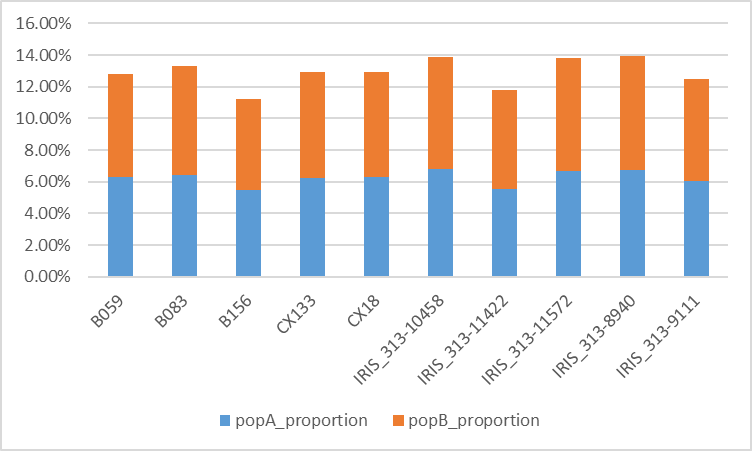
##在此之前需要对popA和popB进行比例的统计,使用awk分sample_name统计,基于受体群体进行,统计popA和popB的比例。